Access CELLxGENE collaboration embeddings (scVI, Geneformer)
This notebook demonstrates basic access to CELLxGENE collaboration embeddings of CELLxGENE Discover Census. Currently, embeddings from scVI and a fine-tuned Geneformer model are maintained by CELLxGENE Discover. There are other CELLxGENE-hosted embeddings contributed by the community to CELLxGENE Discover, find out more about these in the Census model page.
IMPORTANT: This tutorial requires cellxgene-census package version 1.9.1 or later.
Contents
Quick start
Storage format
Query cells and load associated embeddings
⚠️ Note that the Census RNA data includes duplicate cells present across multiple datasets. Duplicate cells can be filtered in or out using the cell metadata variable is_primary_data which is described in the Census schema.
Quick start
CELLxGENE collaboration embeddings can easily be exported into an AnnData as shown below for any slice of Census. This example queries all cells from tongue tissue.
⚠️ Note that Geneformer embeddings are only available for human data
[1]:
import cellxgene_census
emb_names = ["scvi", "geneformer"]
census_version = "2023-12-15"
with cellxgene_census.open_soma(census_version=census_version) as census:
adata = cellxgene_census.get_anndata(
census,
organism="homo_sapiens",
measurement_name="RNA",
obs_value_filter="tissue == 'tongue'",
obs_embeddings=emb_names,
)
[2]:
adata
[2]:
AnnData object with n_obs × n_vars = 372 × 60664
obs: 'soma_joinid', 'dataset_id', 'assay', 'assay_ontology_term_id', 'cell_type', 'cell_type_ontology_term_id', 'development_stage', 'development_stage_ontology_term_id', 'disease', 'disease_ontology_term_id', 'donor_id', 'is_primary_data', 'self_reported_ethnicity', 'self_reported_ethnicity_ontology_term_id', 'sex', 'sex_ontology_term_id', 'suspension_type', 'tissue', 'tissue_ontology_term_id', 'tissue_general', 'tissue_general_ontology_term_id', 'raw_sum', 'nnz', 'raw_mean_nnz', 'raw_variance_nnz', 'n_measured_vars'
var: 'soma_joinid', 'feature_id', 'feature_name', 'feature_length', 'nnz', 'n_measured_obs'
obsm: 'scvi', 'geneformer'
[3]:
adata.obsm
[3]:
AxisArrays with keys: scvi, geneformer
Storage format
Each embedding is encoded as a SOMA SparseNDArray, where:
dimension 0 (
soma_dim_0) encodes the cell (obs)soma_joinidvaluedimension 1 (
soma_dim_1) encodes the embedding feature, and is in the range [0, N) where N is the number of features in the embeddingdata (
soma_data) is float32
The first axis of the embedding array will have the same shape as the corresponding obs DataFrame for the Census build and experiment. The second axis of the embedding will have a shape (0, N) where N is the number of features in the embedding.
Embedding values, while stored as a float32, are precision reduced. Currently they are equivalent to a bfloat16, i.e., have 8 bits of exponent and 7 bits of mantissa.
Query cells and load associated embeddings
This section demonstrates several methods to query cells from the Census by obs metadata, and then fetch embeddings associated with each cell.
Loading embeddings into an AnnData obsm slot
There are two main ways to load CELLxGENE collaboration embeddings into an AnnData.
Via
cellxgene_census.get_anndata().With a lazy query via
ExperimentAxisQuery.
AnnData embeddings via cellxgene_census.get_anndata()
This is the simplest way of getting the embeddings. In this example we create an AnnData for all “central nervous system” cells.
[4]:
import warnings
import cellxgene_census
import scanpy
warnings.filterwarnings("ignore")
census_version = "2023-12-15"
census = cellxgene_census.open_soma(census_version=census_version)
emb_names = ["scvi", "geneformer"]
adata = cellxgene_census.get_anndata(
census,
organism="homo_sapiens",
measurement_name="RNA",
obs_value_filter="tissue_general == 'central nervous system'",
obs_column_names=["cell_type"],
obs_embeddings=emb_names,
)
census.close()
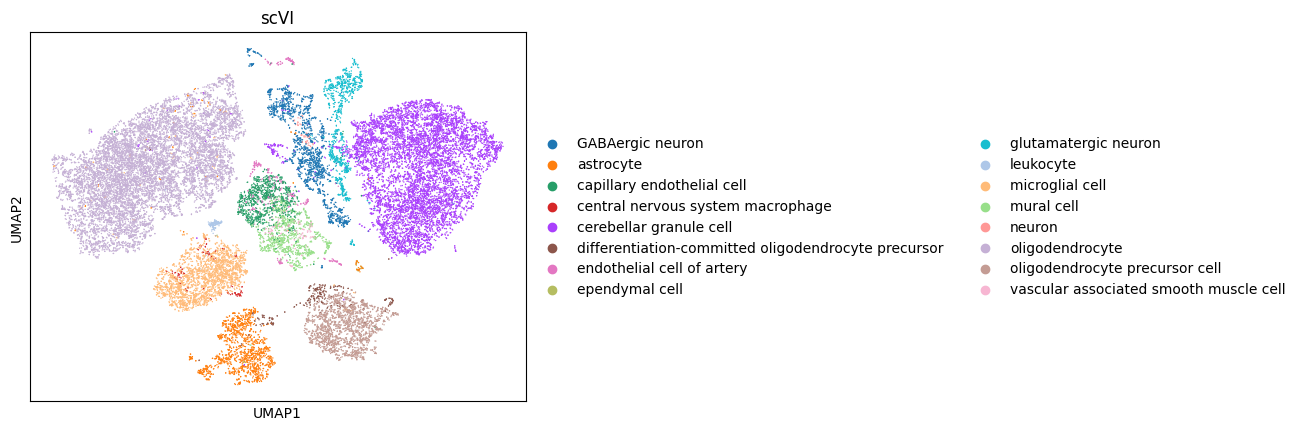
Then we can take a quick look at the embeddings in a 2D scatter plot via UMAP.
[5]:
scanpy.pp.neighbors(adata, use_rep="scvi")
scanpy.tl.umap(adata)
scanpy.pl.umap(adata, color="cell_type", title="scVI")
[6]:
scanpy.pp.neighbors(adata, use_rep="geneformer")
scanpy.tl.umap(adata)
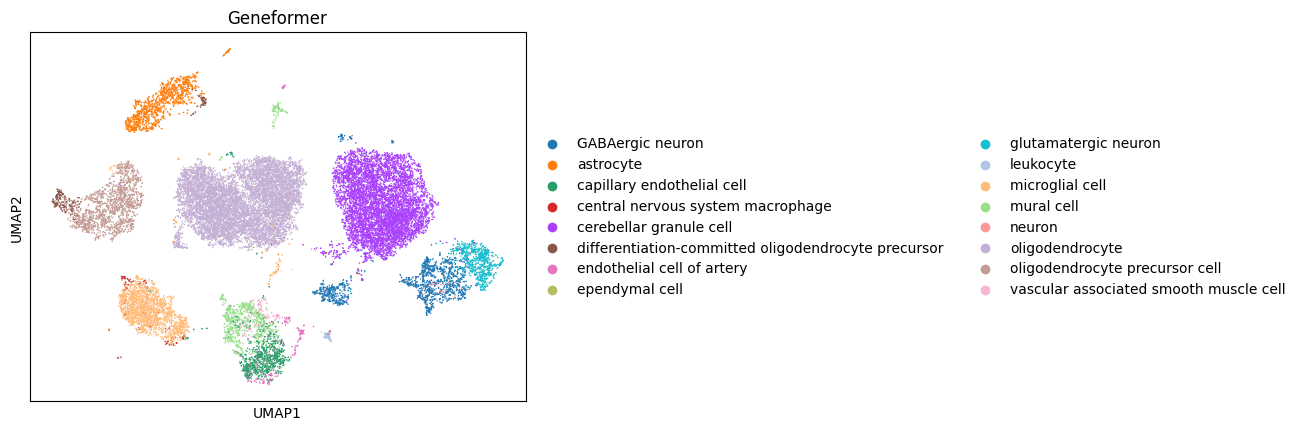
scanpy.pl.umap(adata, color="cell_type", title="Geneformer")
AnnData embeddings via ExperimentAxisQuery
Using an ExperimentAxisQuery to get embeddings into an AnnData has the main advantage of inspecting the query in a lazy manner before loading all data into AnnData.
As a reminder this class offers a lazy interface to query Census based on cell and gene metadata, and provides access to the correspondong expression data, cell/gene metadata, and the embeddings.
Let’s initiate a lazy query with the same filters as the previous example.
[7]:
import cellxgene_census
import scanpy
import tiledbsoma as soma
census_version = "2023-12-15"
organism = "homo_sapiens"
census = cellxgene_census.open_soma(census_version=census_version)
experiment = census["census_data"][organism]
query = experiment.axis_query(
measurement_name="RNA",
obs_query=soma.AxisQuery(value_filter="tissue_general == 'central nervous system'"),
)
Now, before downloading all the data we can take a look at different attributes, for example the number of cells in our query.
[8]:
query.n_obs
[8]:
31780
Then we create an AnnData, retrieve the embeddings and add them to the AnnData’s obsm slot:
[9]:
from cellxgene_census.experimental import get_embedding, get_embedding_metadata_by_name
emb_names = ["scvi", "geneformer"]
adata = query.to_anndata(X_name="raw", column_names={"obs": ["cell_type"]})
for embedding_name in ["scvi", "geneformer"]:
metadata = get_embedding_metadata_by_name(embedding_name, "homo_sapiens", census_version=census_version)
embedding_uri = (
f"s3://cellxgene-contrib-public/contrib/cell-census/soma/{metadata['census_version']}/{metadata['id']}"
)
embedding = get_embedding(metadata["census_version"], embedding_uri, query.obs_joinids().to_numpy())
adata.obsm[embedding_name] = embedding
adata
[9]:
AnnData object with n_obs × n_vars = 31780 × 60664
obs: 'cell_type', 'tissue_general'
var: 'soma_joinid', 'feature_id', 'feature_name', 'feature_length', 'nnz', 'n_measured_obs'
obsm: 'scvi', 'geneformer'
[10]:
adata
[10]:
AnnData object with n_obs × n_vars = 31780 × 60664
obs: 'cell_type', 'tissue_general'
var: 'soma_joinid', 'feature_id', 'feature_name', 'feature_length', 'nnz', 'n_measured_obs'
obsm: 'scvi', 'geneformer'
[11]:
query.close()
census.close()
Load an embedding into a dense NumPy array
To load a embeddinng into a stand-alone numpy array you can select cells from the Census based on obs metadata, then given the resulting cells, use the soma_joinid values to download an embedding, and finally save as a dense NDArray.
Let’s first select cells based on cell metadata.
[12]:
import cellxgene_census
import tiledbsoma as soma
census_version = "2023-12-15"
experiment_name = "homo_sapiens"
census = cellxgene_census.open_soma(census_version=census_version)
obs_df = cellxgene_census.get_obs(
census,
experiment_name,
value_filter="tissue_general == 'central nervous system'",
column_names=["soma_joinid", "cell_type"],
)
Now you can use the soma_joinid values to download the corresponding rows of the embedding matrix via get_embedding.
[13]:
metadata = get_embedding_metadata_by_name("scvi", experiment_name, census_version=census_version)
embedding_uri = f"s3://cellxgene-contrib-public/contrib/cell-census/soma/{metadata['census_version']}/{metadata['id']}"
embedding = get_embedding(metadata["census_version"], embedding_uri, obs_df.soma_joinid.to_numpy())
And now we have a dense matrix with the embedding data.
[14]:
embedding
[14]:
array([[-6.17187500e-01, 3.82995605e-03, -7.50000000e-01, ...,
7.50000000e-01, 9.39941406e-03, 2.71606445e-03],
[ 3.39843750e-01, 4.71115112e-04, -8.32031250e-01, ...,
8.00781250e-01, -7.55310059e-04, 8.85009766e-03],
[ 4.10156250e-01, -2.42614746e-03, -5.00000000e-01, ...,
9.45312500e-01, -2.53295898e-03, 1.12915039e-02],
...,
[ 3.84765625e-01, -1.54113770e-03, -1.94531250e+00, ...,
-2.38281250e-01, -1.03149414e-02, 2.28881836e-03],
[ 8.94531250e-01, -7.14111328e-03, 5.78125000e-01, ...,
-1.15234375e-01, -2.39562988e-03, 2.42614746e-03],
[ 6.79687500e-01, -8.48388672e-03, 1.45312500e+00, ...,
-1.19628906e-01, 6.62803650e-05, -1.88446045e-03]], dtype=float32)
[16]:
embedding.shape
[16]:
(31780, 50)
[17]:
query.close()
census.close()