Access CELLxGENE-hosted embeddings
This notebook demonstrates basic access to CELLxGENE-hosted embeddings of the Census. CELLxGENE-hosted embeddings have been contributed by the community, CELLxGENE Discover does not actively maintain or update them. Find out more about these in the Census model page.
IMPORTANT: This tutorial requires cellxgene-census package version 1.9.1 or later.
Contents
Background
Quick start
Query cells and load associated embeddings
Load embeddings and fetch associated Census data
Embedding metadata
⚠️ Note that the Census RNA data includes duplicate cells present across multiple datasets. Duplicate cells can be filtered in or out using the cell metadata variable
is_primary_datawhich is described in the Census schema.
Background
This notebook demonstrates access to CELLxGENE-hosted embeddings of the Census. The Census has multiple releases, named by a census_version, which normally looks like an ISO date, e.g., 2023-02-01. A CELLxGENE-hosted embedding is a 2D sparse matrix of cell embeddings for a given census version, encoded as a SOMA SparseNDArray.
⚠️ Note that embeddings may be available for one or both organisms, see the Census model page for the latest availability.
⚠️ IMPORTANT: embeddings are only meaningful in the context of the Census from which they were created. Each embedding contains a metadata field indicating the source Census, suitable for confirming embedding lineage.
Quick start
The easiest way to access Census CELLxGENE-hosted embeddings is by calling the get_anndata function with an obs_embeddings or var_embeddings parameter.
Let’s start by exploring what embeddings are available:
[1]:
from cellxgene_census.experimental import get_all_available_embeddings
CENSUS_VERSION = "2023-12-15"
for e in get_all_available_embeddings(CENSUS_VERSION):
print(f"{e['embedding_name']:15} {e['experiment_name']:15} {e['data_type']:15}")
scvi mus_musculus obs_embedding
scvi homo_sapiens obs_embedding
geneformer homo_sapiens obs_embedding
scgpt homo_sapiens obs_embedding
uce homo_sapiens obs_embedding
uce mus_musculus obs_embedding
nmf homo_sapiens obs_embedding
nmf homo_sapiens var_embedding
These can also be viewed on the CELLxGENE Census Models page.
For this example, we’ll use scgpt. We can call get_anndata with the obs_embeddings parameter:
[2]:
import cellxgene_census
from cellxgene_census.experimental import get_embedding
CENSUS_VERSION = "2023-12-15"
with cellxgene_census.open_soma(census_version=CENSUS_VERSION) as census:
adata = cellxgene_census.get_anndata(
census,
organism="homo_sapiens",
measurement_name="RNA",
obs_value_filter="tissue == 'tongue'",
obs_embeddings=["scgpt"],
)
And now you can use these data for downstream analysis.
[3]:
adata
[3]:
AnnData object with n_obs × n_vars = 372 × 60664
obs: 'soma_joinid', 'dataset_id', 'assay', 'assay_ontology_term_id', 'cell_type', 'cell_type_ontology_term_id', 'development_stage', 'development_stage_ontology_term_id', 'disease', 'disease_ontology_term_id', 'donor_id', 'is_primary_data', 'self_reported_ethnicity', 'self_reported_ethnicity_ontology_term_id', 'sex', 'sex_ontology_term_id', 'suspension_type', 'tissue', 'tissue_ontology_term_id', 'tissue_general', 'tissue_general_ontology_term_id', 'raw_sum', 'nnz', 'raw_mean_nnz', 'raw_variance_nnz', 'n_measured_vars'
var: 'soma_joinid', 'feature_id', 'feature_name', 'feature_length', 'nnz', 'n_measured_obs'
obsm: 'scgpt'
[4]:
adata.obsm
[4]:
AxisArrays with keys: scgpt
⚠️ IMPORTANT: get_embedding will fill in missing values in the embedding matrix with NaN, therefore missing cells are represented with rows where all values are NaN. To learn more use help(get_embedding).
Storage format
Each embedding is encoded as a SOMA SparseNDArray, where:
dimension 0 (
soma_dim_0) encodes the cell (obs)soma_joinidvaluedimension 1 (
soma_dim_1) encodes the embedding feature, and is in the range [0, N) where N is the number of featues in the embeddingdata (
soma_data) is float32
⚠️ IMPORTANT: CELLxGENE-hosted embeddings may embed a subset of the cells in any given Census version. If a cell has an embedding, it will be explicitly stored in the sparse array, even if the embedding value is zero. In other words, missing array values values imply that the cell was not embedded, whereas zero valued embeddings are explicitly stored. Put another way, the nnz of the embedding array indicate the number of embedded cells, not the number of non-zero values.
The first axis of the embedding array will have the same shape as the corresponding obs DataFrame for the Census build and experiment. The second axis of the embedding will have a shape (0, N) where N is the number of features in the embedding.
Embedding values, while stored as a float32, are precision reduced. Currently they are equivalent to a bfloat16, i.e., have 8 bits of exponent and 7 bits of mantissa.
Query cells and load associated embeddings
This section demonstrates two methods to query cells from the Census by obs metadata, and then fetch CELLxGENE-hosted embeddings associated with each cell.
Load an embedding into an AnnData
obsmslotLoad an embedding into a dense NumPy array
Let’s first do a few imports and utility functions used throughout this notebook.
[5]:
# A few imports and utility functions used throughout this notebook
import warnings
import cellxgene_census
import numpy as np
import scanpy
import tiledbsoma as soma
from cellxgene_census.experimental import get_embedding_metadata
warnings.filterwarnings("ignore")
# The Census version to utilize
CENSUS_VERSION = "2023-12-15"
EXPERIMENT_NAME = "homo_sapiens"
MEASUREMENT_NAME = "RNA"
# The location of the embedding. See <https://cellxgene.cziscience.com/census-models>
# for available CELLxGENE-hosted embeddings.
EMBEDDING_URI = "s3://cellxgene-contrib-public/contrib/cell-census/soma/2023-12-15/CxG-contrib-1/"
Load an embedding into an AnnData obsm slot
There are two main ways to load hosted embeddings into an AnnData.
Via
cellxgene_census.get_anndata(), followed by merging embeddings.With a lazy query via
ExperimentAxisQuery, followed by merging embeddings.
AnnData embeddings via cellxgene_census.get_anndata()
This is the simplest way of getting the embeddings. In this example we create an AnnData for all “central nervous system” cells, and use the obs_embeddings parameter to add scGPT embeddings to the obsm slot.
[6]:
with cellxgene_census.open_soma(census_version=CENSUS_VERSION) as census:
adata = cellxgene_census.get_anndata(
census,
organism=EXPERIMENT_NAME,
measurement_name=MEASUREMENT_NAME,
obs_value_filter="tissue_general == 'central nervous system'",
obs_column_names=["cell_type", "soma_joinid"],
obs_embeddings=["scgpt"],
)
⚠️ IMPORTANT: get_embedding will fill in missing values in the embedding matrix with NaN, therefore missing cells are represented with rows where all values are NaN. To learn more use help(get_embedding).
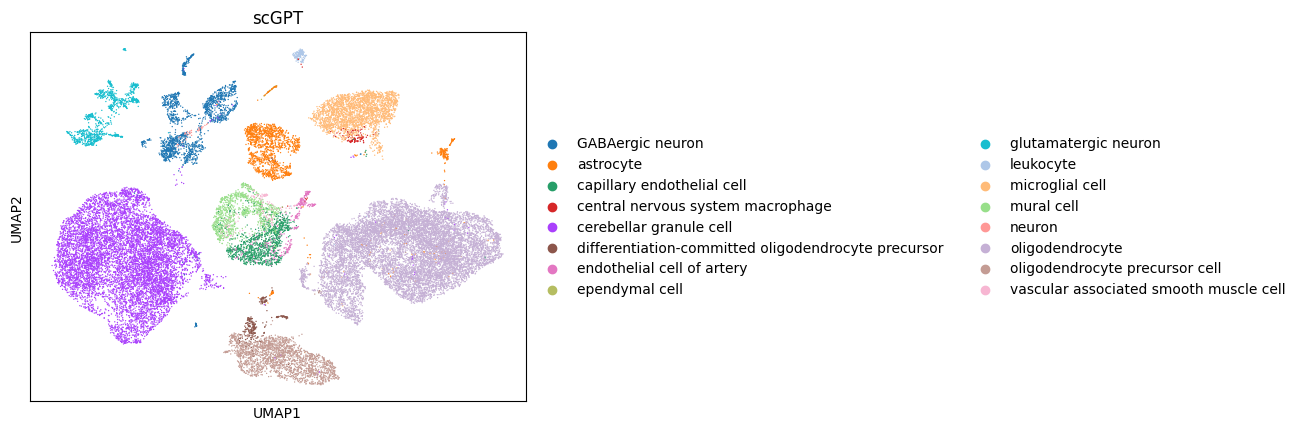
Then we can take a quick look at the embeddings in a 2D scatter plot via UMAP.
[7]:
scanpy.pp.neighbors(adata, use_rep="scgpt")
scanpy.tl.umap(adata)
scanpy.pl.umap(adata, color="cell_type", title="scGPT")
AnnData embeddings via ExperimentAxisQuery
Using an ExperimentAxisQuery to get embeddings into an AnnData has the main advantage of inspecting the query in a lazy manner before loading all data into AnnData.
As a reminder this class offers a lazy interface to query Census based on cell and gene metadata, and provides access to the correspondong expression data, and cell/gene metadata.
Let’s initiate a lazy query with the same filters as the previous example.
[8]:
census = cellxgene_census.open_soma(census_version=CENSUS_VERSION)
experiment = census["census_data"][EXPERIMENT_NAME]
query = experiment.axis_query(
measurement_name="RNA",
obs_query=soma.AxisQuery(value_filter="tissue_general == 'central nervous system'"),
)
Now, before downloading all the data we can take a look at different attributes, for example the number of cells in our query.
[9]:
query.n_obs
[9]:
31780
And for example grab all the soma_joinid values for the cells in this query.
[10]:
soma_joinids = query.obs_joinids().to_numpy()
Then create an AnnData.
[11]:
adata = query.to_anndata(X_name="raw", column_names={"obs": ["cell_type"]})
And finally add the embedding matrix via get_embedding for the cells of the query using the soma_joinid values.
[12]:
adata.obsm["scgpt"] = get_embedding(
census_version=CENSUS_VERSION,
embedding_uri=EMBEDDING_URI,
obs_soma_joinids=soma_joinids,
)
⚠️ IMPORTANT: get_embedding will fill in missing values in the embedding matrix with NaN, therefore missing cells are represented with rows where all values are NaN. To learn more use help(get_embedding).
[13]:
adata
[13]:
AnnData object with n_obs × n_vars = 31780 × 60664
obs: 'cell_type', 'tissue_general'
var: 'soma_joinid', 'feature_id', 'feature_name', 'feature_length', 'nnz', 'n_measured_obs'
obsm: 'scgpt'
[14]:
query.close()
census.close()
Load an embedding into a dense NumPy array
To load a embeddinng into a stand-alone numpy array you can select cells from the Census based on obs metadata, then given the resulting cells, use the soma_joinid values to download an embedding, and finally save as a dense NDArray.
Let’s first select cells based on cell metadata
[15]:
census = cellxgene_census.open_soma(census_version=CENSUS_VERSION)
obs_df = cellxgene_census.get_obs(
census,
EXPERIMENT_NAME,
value_filter="tissue_general == 'exocrine gland'",
column_names=["soma_joinid", "cell_type"],
)
Now you can use the soma_joinid values to download the corresponding rows of the embedding matrix via get_embedding.
[16]:
embeddings = get_embedding(CENSUS_VERSION, EMBEDDING_URI, obs_df.soma_joinid.to_numpy())
embeddings[0:3, 0:4]
[16]:
array([[-0.00506592, -0.01348877, -0.03173828, -0.02331543],
[ 0.02404785, -0.02441406, -0.00595093, -0.0065918 ],
[ 0.00070572, 0.00187683, -0.04663086, -0.04614258]],
dtype=float32)
⚠️ IMPORTANT: get_embedding will fill in missing values in the embedding matrix with NaN, therefore missing cells are represented with rows where all values are NaN.
[17]:
embeddings.shape
[17]:
(115722, 512)
Load embeddings and fetch associated Census data
This section describes a more advanced use case. Here we showcase how to load large slices of an embeding matrix, and then append cell metadata to them.
The method starts with the loaded embedding, and for each embedded cell loads metadata or X data.
[18]:
# Load a portion of the embedding (caution: embeddings can be quite large)
import cellxgene_census
import tiledbsoma as soma
# Fetch first 500_000 joinids from the embedding.
# NOTE: will fail if the there are no cells embedded within this obs joinid range
embedding_slice = (slice(500_000),)
emb_data = []
emb_joinids = []
ctx = {"vfs.s3.region": "us-west-2", "vfs.s3.no_sign_request": True}
with soma.open(EMBEDDING_URI, context=soma.SOMATileDBContext(tiledb_config=ctx)) as E:
# read embedding and obs joinids for each embedded cell
for d, (obs_joinids, _) in (
E.read(coords=embedding_slice).blockwise(axis=0, size=2**20, reindex_disable_on_axis=1).scipy()
):
embedding_presence_mask = d.getnnz(axis=1) != 0
emb_joinids.append(obs_joinids[embedding_presence_mask])
emb_data.append(d[embedding_presence_mask, :].toarray())
# concat
embedding_data = np.vstack(emb_data)
embedding_joinids = np.concatenate(emb_joinids)
# Load the associated metadata - in this case, obs.suspension_type
with cellxgene_census.open_soma(census_version=CENSUS_VERSION) as census:
experiment = census["census_data"][EXPERIMENT_NAME]
with experiment.axis_query(
measurement_name=MEASUREMENT_NAME,
obs_query=soma.AxisQuery(coords=(embedding_joinids,)),
) as query:
obs_df = query.obs(column_names=["soma_joinid", "suspension_type"]).concat().to_pandas()
# Display the first few cells, and the first few columns of their embeddings
display(obs_df[0:3])
display(embedding_data[0:3, 0:4])
| soma_joinid | suspension_type | |
|---|---|---|
| 0 | 0 | nucleus |
| 1 | 1 | nucleus |
| 2 | 2 | nucleus |
array([[-0.00762939, -0.00076675, -0.00047874, -0.03588867],
[ 0.00405884, -0.00239563, 0.00982666, -0.00946045],
[ 0.00473022, 0.0135498 , -0.01049805, -0.03051758]],
dtype=float32)
Embedding Metadata
Each embedding contains descriptive information stored in the SOMA metadata slot, encoded as a JSON string. This metadata includes:
census_version - the Census which is embedded. It is critical to confirm this matches the Census in use, or the embeddings will be meaningless.
experiment_name - the Census experiment embedded, e.g.,
homo_sapiensormus_musculus.measurement_name - the Census measurement embedded, e.g.,
RNA
There are a variety of other metadata values documented here.
The following example demonstrates how to access and decode the metadata into a Python dictionary.
[19]:
embedding_metadata = get_embedding_metadata(EMBEDDING_URI)
embedding_metadata
[19]:
{'id': 'CxG-contrib-1',
'title': 'scGPT: Towards Building a Foundation Model for Single-Cell Multi-omics Using Generative AI',
'description': 'Utilizing the burgeoning single-cell sequencing data, we have pioneered the construction of a foundation model for single-cell biology, scGPT, which is based on generative pre-trained transformer across a repository of over 33 million cells. Our findings illustrate that scGPT, a generative pre-trained transformer, effectively distills critical biological insights concerning genes and cells. Through the further adaptation of transfer learning, scGPT can be optimized to achieve superior performance across diverse downstream applications.',
'primary_contact': {'name': 'Bo Wang',
'email': 'bowang@vectorinstitute.ai',
'affiliation': 'Bo Wang Lab, University of Toronto'},
'additional_contacts': [],
'DOI': '10.1101/2023.04.30.538439',
'additional_information': '',
'model_link': 'https://github.com/bowang-lab/scGPT',
'data_type': 'obs_embedding',
'census_version': '2023-12-15',
'experiment_name': 'homo_sapiens',
'measurement_name': 'RNA',
'n_embeddings': 62998417,
'n_features': 512,
'submission_date': '2023-11-09'}
A common use case for accessing embedding metadata is the prevention of nonsense results, which will occur when the embeddings were not derived from the Census or experiment in use.
This demonstrates an easy way to grab the embedding metadata and confirm that it matches your expectations.
[20]:
embedding_metadata = get_embedding_metadata(EMBEDDING_URI)
assert embedding_metadata["census_version"] == CENSUS_VERSION
assert embedding_metadata["experiment_name"] == EXPERIMENT_NAME
assert embedding_metadata["measurement_name"] == MEASUREMENT_NAME
display("all good!")
'all good!'