Geneformer for cell class prediction and data projection
This notebook provides examples to utilize the CELLxGENE collaboration fine-tuned Geneformer model with user data. For more information on the model please refer to the Census model page.
IMPORTANT: This tutorial requires cellxgene-census package version 1.9.1 or later.
Contents
Requirements.
Preparing data and model.
Using the Geneformer fine-tuned model for cell subclass inference.
Using the Geneformer fine-tuned model for data projection.
⚠️ Note “cell subclass” is a high-level grouping of cell types as annotated in CELLxGENE Discover via the CL ontology see [https://cellxgene.cziscience.com/collections](https://cellxgene.cziscience.com/collections
⚠️ Note that the Census RNA data includes duplicate cells present across multiple datasets. Duplicate cells can be filtered in or out using the cell metadata variable
is_primary_datawhich is described in the Census schema.
Requirements
System requirements
To run this notebook the following are required:
Unix system.
A system with one or more GPUs is highly recommended.
Geneformer Python package and its dependencies.
CELLxGENE Census package.
Downloading example data
Throughout the notebook the 10X PBMC 3K dataset will be used, you can download it via the following shell commands.
[1]:
!mkdir -p data
!wget -nv -O data/pbmc3k_filtered_gene_bc_matrices.tar.gz http://cf.10xgenomics.com/samples/cell-exp/1.1.0/pbmc3k/pbmc3k_filtered_gene_bc_matrices.tar.gz
!tar -xzf data/pbmc3k_filtered_gene_bc_matrices.tar.gz -C data/
2024-05-10 17:53:27 URL:https://cf.10xgenomics.com/samples/cell-exp/1.1.0/pbmc3k/pbmc3k_filtered_gene_bc_matrices.tar.gz [7621991/7621991] -> "data/pbmc3k_filtered_gene_bc_matrices.tar.gz" [1]
Downloading the fine-tuned Geneformer model
The model is currently hosted in S3, you can find out more deatails in the Census model page.
Additional information, including its S3 URI, is also included in the metadata of the corresponding embeddings inside Census. These metadata can be obtained as follows.
[2]:
import cellxgene_census
import cellxgene_census.experimental
census_version = "2023-12-15"
organism = "homo_sapiens"
census = cellxgene_census.open_soma(census_version=census_version)
geneformer_info = cellxgene_census.experimental.get_embedding_metadata_by_name(
embedding_name="geneformer",
organism=organism,
census_version=census_version,
)
[3]:
geneformer_info["model_link"]
[3]:
's3://cellxgene-contrib-public/models/geneformer/2023-12-15/homo_sapiens/fined-tuned-model/'
And we can download it via the AWS CLI.
[4]:
!aws s3 sync --no-sign-request --no-progress --only-show-errors s3://cellxgene-contrib-public/models/geneformer/2023-12-15/homo_sapiens/fined-tuned-model/ ./fine_tuned_geneformer
Importing required packages
Finally all the required packages are loaded.
[5]:
import warnings
warnings.filterwarnings("ignore")
import json
import os
import cellxgene_census
import datasets
import numpy as np
import scanpy as sc
from geneformer import (
DataCollatorForCellClassification,
EmbExtractor,
TranscriptomeTokenizer,
)
from transformers import BertForSequenceClassification, Trainer
Preparing data and model
Preparing single-cell data
Let’s load the test data. In preparation to use with Geneformer we do the following:
Set the index as the ENSEMBL gene ID and stores it in the
obscolumn"ensembl_id"e.g.
ENSG00000139618(without a version number suffix)
Add read counts to the
obscolumn"n_counts"Add an ID column to be used for joining later in the
obscolumn"joinid"
Then we write the resulting H5AD file to disk.
[6]:
adata = sc.read_10x_mtx("data/filtered_gene_bc_matrices/hg19/", var_names="gene_ids")
adata.var["ensembl_id"] = adata.var.index
adata.obs["n_counts"] = adata.X.sum(axis=1)
adata.obs["joinid"] = list(range(adata.n_obs))
h5ad_dir = "./data/h5ad/"
if not os.path.exists(h5ad_dir):
os.makedirs(h5ad_dir)
adata.write(h5ad_dir + "pbmcs.h5ad")
Now we can tokenize the test data using Geneformer’s tokenizer, while keeping track of "joinid" for future joining.
[7]:
token_dir = "data/tokenized_data/"
if not os.path.exists(token_dir):
os.makedirs(token_dir)
tokenizer = TranscriptomeTokenizer(custom_attr_name_dict={"joinid": "joinid"})
tokenizer.tokenize_data(
data_directory=h5ad_dir,
output_directory=token_dir,
output_prefix="pbmc",
file_format="h5ad",
)
Tokenizing data/h5ad/pbmcs.h5ad
data/h5ad/pbmcs.h5ad has no column attribute 'filter_pass'; tokenizing all cells.
Creating dataset.
Preparing data from model
Then let’s fetch the mapping dictionary between Geneformer IDs and the associated cell subclass labels. This information is stored along the fine-tuned model.
[8]:
model_dir = "./fine_tuned_geneformer/"
label_mapping_dict_file = os.path.join(model_dir, "label_to_cell_subclass.json")
with open(label_mapping_dict_file) as fp:
label_mapping_dict = json.load(fp)
This dictionary contains all the possible cell labels available for the model, and the predictions on the section below will use these labels.
[9]:
label_mapping_dict
[9]:
{'0': 'B cell',
'1': 'BEST4+ intestinal epithelial cell, human',
'2': 'CD4-positive, alpha-beta T cell',
'3': 'CD8-positive, alpha-beta T cell',
'4': 'CNS neuron (sensu Vertebrata)',
'5': 'GABAergic neuron',
'6': 'T cell',
'7': 'abnormal cell',
'8': 'adventitial cell',
'9': 'animal cell',
'10': 'bone cell',
'11': 'cardiocyte',
'12': 'cell of skeletal muscle',
'13': 'ciliated cell',
'14': 'columnar/cuboidal epithelial cell',
'15': 'connective tissue cell',
'16': 'contractile cell',
'17': 'defensive cell',
'18': 'dendritic cell',
'19': 'duct epithelial cell',
'20': 'ecto-epithelial cell',
'21': 'ectodermal cell',
'22': 'endo-epithelial cell',
'23': 'endocrine cell',
'24': 'endothelial cell',
'25': 'epithelial cell',
'26': 'epithelial cell of lung',
'27': 'epithelial cell of pancreas',
'28': 'epithelial cell of urethra',
'29': 'eukaryotic cell',
'30': 'exocrine cell',
'31': 'fat cell',
'32': 'fibroblast',
'33': 'germ cell',
'34': 'glandular epithelial cell',
'35': 'glial cell',
'36': 'glutamatergic neuron',
'37': 'hematopoietic cell',
'38': 'hematopoietic precursor cell',
'39': 'hepatocyte',
'40': 'inflammatory cell',
'41': 'interneuron',
'42': 'interstitial cell',
'43': 'ionocyte',
'44': 'kidney cell',
'45': 'kidney epithelial cell',
'46': 'leukocyte',
'47': 'lymphocyte',
'48': 'macrophage',
'49': 'male germ cell',
'50': 'mammary gland epithelial cell',
'51': 'mesenchymal cell',
'52': 'meso-epithelial cell',
'53': 'mesodermal cell',
'54': 'monocyte',
'55': 'motor neuron',
'56': 'mural cell',
'57': 'muscle cell',
'58': 'myeloid cell',
'59': 'myofibroblast cell',
'60': 'neoplastic cell',
'61': 'neural cell',
'62': 'neuron',
'63': 'neuron associated cell',
'64': 'non-terminally differentiated cell',
'65': 'ovarian surface epithelial cell',
'66': 'pericyte',
'67': 'phagocyte',
'68': 'pigment cell',
'69': 'precursor cell',
'70': 'primary cultured cell',
'71': 'primordial germ cell',
'72': 'progenitor cell',
'73': 'salivary gland cell',
'74': 'sebaceous gland cell',
'75': 'secretory cell',
'76': 'sensory neuron',
'77': 'seromucus secreting cell',
'78': 'somatic cell',
'79': 'squamous epithelial cell',
'80': 'stem cell',
'81': 'stratified epithelial cell',
'82': 'stromal cell',
'83': 'supporting cell',
'84': 'transit amplifying cell',
'85': 'transitional epithelial cell',
'86': 'trophoblast cell',
'87': 'vertebrate lens cell'}
Using the Geneformer fine-tuned model for cell subclass inference
Loading tokenized data
Let’s load the tokenized test data.
[10]:
dataset = datasets.load_from_disk(token_dir + "pbmc.dataset")
dataset
[10]:
Dataset({
features: ['input_ids', 'joinid', 'length'],
num_rows: 2700
})
We add a dummy cell metadata column "label" needed for Geneformer to make predictions.
[11]:
dataset
dataset = dataset.add_column("label", [0] * len(dataset))
Performing inference of cell subclass
Now we can load the model and run the inference workflow.
⚠️ Note, this step will be slow with CPUs, a machine with one GPU is recommended
[12]:
# reload pretrained model
model = BertForSequenceClassification.from_pretrained(model_dir)
# create the trainer
trainer = Trainer(model=model, data_collator=DataCollatorForCellClassification())
# use trainer
predictions = trainer.predict(dataset)
And finally we select the most likely cell class based on the probability vector from the predictions of each cell in our test data.
[13]:
predicted_label_ids = np.argmax(predictions.predictions, axis=1)
predicted_logits = [predictions.predictions[i][predicted_label_ids[i]] for i in range(len(predicted_label_ids))]
predicted_labels = [label_mapping_dict[str(i)] for i in predicted_label_ids]
Inspecting inference results
Then we add the prediction back to our loaded AnnData test dataset.
[14]:
adata.obs["predicted_cell_subclass"] = predicted_labels
adata.obs["predicted_cell_subclass_probability"] = np.exp(predicted_logits) / (1 + np.exp(predicted_logits))
And it’s ready for inspecting the predictions. Let’s visualize the predictions on the UMAP space, the following is a basic processing workflow to derive a UMAP representation, of the data.
[15]:
sc.pp.normalize_total(adata, target_sum=1e4)
sc.pp.log1p(adata)
sc.pp.highly_variable_genes(adata, min_mean=0.0125, max_mean=3, min_disp=0.5)
adata = adata[:, adata.var.highly_variable]
sc.pp.scale(adata, max_value=10)
sc.tl.pca(adata, svd_solver="arpack")
sc.pp.neighbors(adata, n_neighbors=10, n_pcs=40)
sc.tl.umap(adata)
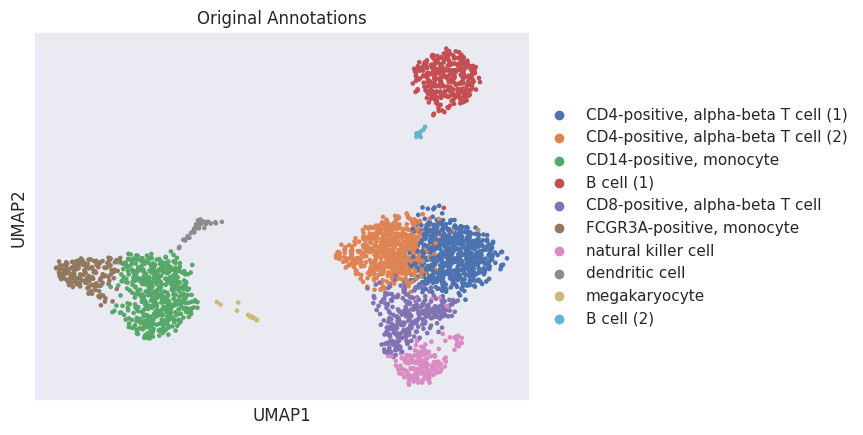
Let’s also add the original cell type annotations as obtained in Scapy’s annotation tutorial of the same data.
[16]:
sc.tl.leiden(adata)
original_cell_types = [
"CD4-positive, alpha-beta T cell (1)",
"CD4-positive, alpha-beta T cell (2)",
"CD14-positive, monocyte",
"B cell (1)",
"CD8-positive, alpha-beta T cell",
"FCGR3A-positive, monocyte",
"natural killer cell",
"dendritic cell",
"megakaryocyte",
"B cell (2)",
]
adata.rename_categories("leiden", original_cell_types)
These are the original annotations.
[17]:
sc.pl.umap(adata, color="leiden", title="Original Annotations")
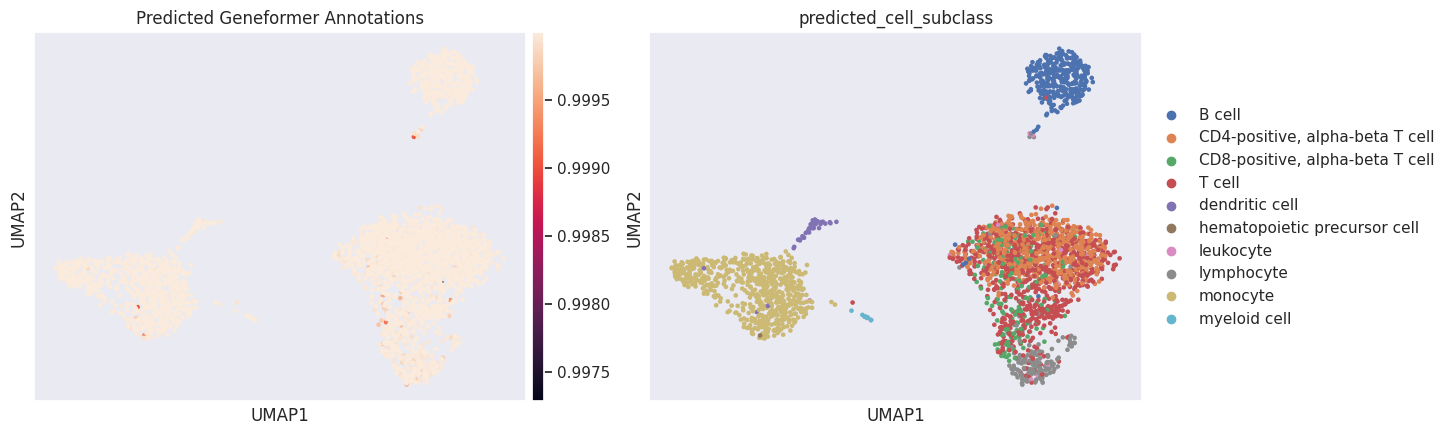
And these are the predicted annotations.
[18]:
sc.pl.umap(
adata,
color=["predicted_cell_subclass_probability", "predicted_cell_subclass"],
title="Predicted Geneformer Annotations",
)
WARNING: The title list is shorter than the number of panels. Using 'color' value instead for some plots.
Using the Geneformer fine-tuned model for data projection
Generating Geneformer embeddings for 10X PBMC 3K data
To project new data, for example the 10X PBMC 3K data, into the Census embedding space from Geneformer’s fine-tune model, we can use EmbExtractor from the Geneformer package as follows.
We first need to get the number of categories (cell subclasses) present in the model.
[19]:
n_classes = len(label_mapping_dict)
Then we can run the EmbExtractor, which randomize the cells during the process and thus we keep track of "joinid".
⚠️ Note, this step will be slow with CPUs, a machine with one GPU is recommended
[20]:
output_dir = "data/geneformer_embeddings"
if not os.path.exists(output_dir):
os.makedirs(output_dir)
embex = EmbExtractor(
model_type="CellClassifier",
num_classes=n_classes,
max_ncells=None,
emb_label=["joinid"],
emb_layer=0,
forward_batch_size=30,
nproc=8,
)
embs = embex.extract_embs(
model_directory=model_dir,
input_data_file=token_dir + "pbmc.dataset",
output_directory=output_dir,
output_prefix="emb",
)
Then we simply re-order the embeddings based on "joinid" and then merge them to the original AnnData
[21]:
embs = embs.sort_values("joinid")
adata.obsm["geneformer"] = embs.drop(columns="joinid").to_numpy()
Let’s take a look at these Geneformer embeddings in a UMAP representation
[22]:
sc.pp.neighbors(adata, n_neighbors=10, n_pcs=40, use_rep="geneformer")
sc.tl.umap(adata)
[23]:
sc.pl.umap(adata, color="predicted_cell_subclass", title="10X PBMC 3K in Geneformer")
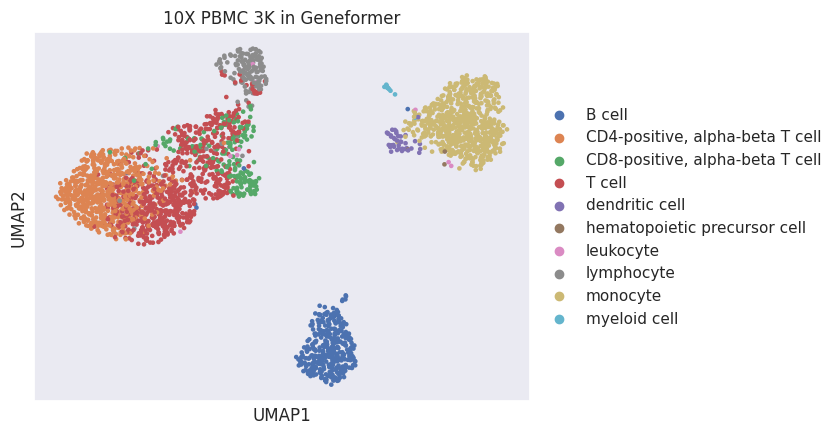
Joining Geneformer embeddings from 10X PBMC 3K data with other Census datasets
There are multiple datasets in Census from PBMCs, and all human Census data has pre-calculated Geneformer embeddings, so now we can join the embeddings we generated above from the 10X PBMC 3K dataset with Census data.
Let’s grab a few PBMC datasets from Census and request the Geneformer embeddings.
[24]:
census = cellxgene_census.open_soma(census_version="2023-12-15")
# Some PBMC data from these collections
# 1. https://cellxgene.cziscience.com/collections/c697eaaf-a3be-4251-b036-5f9052179e70
# 2. https://cellxgene.cziscience.com/collections/f2a488bf-782f-4c20-a8e5-cb34d48c1f7e
dataset_ids = [
"fa8605cf-f27e-44af-ac2a-476bee4410d3",
"3c75a463-6a87-4132-83a8-c3002624394d",
]
adata_census = cellxgene_census.get_anndata(
census=census,
measurement_name="RNA",
organism="Homo sapiens",
obs_value_filter=f"dataset_id in {dataset_ids}",
obs_embeddings=["geneformer"],
)
To simplify let’s select the genes that are also present in the 10X PBMC 3K dataset.
[25]:
adata_census.var_names = adata_census.var["feature_id"]
shared_genes = list(set(adata.var_names) & set(adata_census.var_names))
adata_census = adata_census[:, shared_genes]
And take a subset of these cells, let’s take 3K cells to match the size of the test data.
[26]:
index_subset = np.random.choice(adata_census.n_obs, size=3000, replace=False)
adata_census = adata_census[index_subset, :]
Now we can join these Census data to the 10X PBMC 3K data
[27]:
adata_census.obs["dataset"] = "Census - " + adata_census.obs["dataset_id"].astype(str)
adata.obs["dataset"] = "10X PBMC 3K"
adata.obs["cell_type"] = "Predicted - " + adata.obs["predicted_cell_subclass"].astype(str)
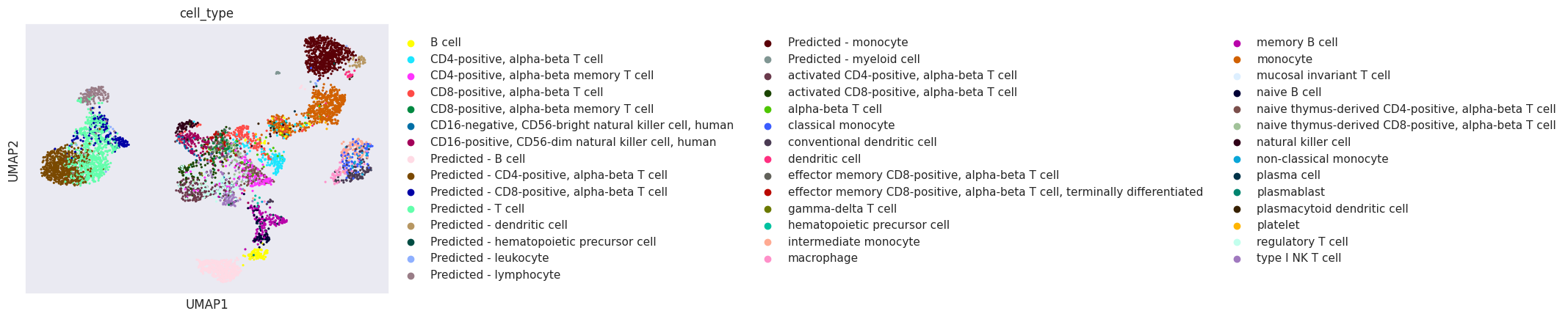
adata_joined = sc.concat([adata, adata_census], join="outer", label="batch")
Let’s now inspect all of the cells in the UMAP space.
[28]:
sc.pp.neighbors(adata_joined, n_neighbors=10, n_pcs=40, use_rep="geneformer")
sc.tl.umap(adata_joined)
[29]:
sc.pl.umap(adata_joined, color="dataset")

[30]:
sc.pl.umap(adata_joined, color="cell_type")