Querying data using the gget cellxgene module
By Laura Luebbert, lauralubbert@gmail.com.
gget is a free, open-source command-line tool and Python package that enables efficient querying of genomic databases. gget consists of a collection of separate but interoperable modules, each designed to facilitate one type of database querying in a single line of code.
The gget cellxgene module builds on the CZ CELLxGENE Discover Census to query data from CZ CELLxGENE Discover. This notebook briefly introduces the gget cellxgene module by providing one simple example for each supported query type.
If you use gget cellxgene in a publication, please cite gget in addition to citing CZ CELLxGENE.
You can also open this notebook in Google Colab.
Contents
Install gget.
Fetch an AnnData object by selecting gene(s), tissue(s) and cell type(s).
Plot a dot plot similar to those shown on the CZ CELLxGENE Discover Gene Expression.
Fetch only cell metadata (corresponds to AnnData.obs).
Use gget cellxgene from the command line.
Install gget and set up cellxgene module
[1]:
# The cellxgene module was added to gget in version 0.25.7
!pip install -q gget >=0.25.7
import gget
gget.setup("cellxgene")
[notice] A new release of pip available: 22.3.1 -> 23.2.1
[notice] To update, run: pip install --upgrade pip
Fri Jul 28 16:16:17 2023 INFO Installing cellxgene-census package (requires pip).
Fri Jul 28 16:16:22 2023 INFO cellxgene_census installed succesfully.
[2]:
# Display all options of the cellxgene gget module
help(gget.cellxgene)
Help on function cellxgene in module gget.gget_cellxgene:
cellxgene(species='homo_sapiens', gene=None, ensembl=False, column_names=['dataset_id', 'assay', 'suspension_type', 'sex', 'tissue_general', 'tissue', 'cell_type'], meta_only=False, tissue=None, cell_type=None, development_stage=None, disease=None, sex=None, is_primary_data=True, dataset_id=None, tissue_general_ontology_term_id=None, tissue_general=None, assay_ontology_term_id=None, assay=None, cell_type_ontology_term_id=None, development_stage_ontology_term_id=None, disease_ontology_term_id=None, donor_id=None, self_reported_ethnicity_ontology_term_id=None, self_reported_ethnicity=None, sex_ontology_term_id=None, suspension_type=None, tissue_ontology_term_id=None, census_version='stable', verbose=True, out=None)
Query data from CZ CELLxGENE Discover (https://cellxgene.cziscience.com/) using the
CZ CELLxGENE Discover Census (https://github.com/chanzuckerberg/cellxgene-census).
NOTE: Querying large datasets requires a large amount of RAM. Use the cell metadata attributes
to define the (sub)dataset of interest.
The CZ CELLxGENE Discover Census recommends >16 GB of memory and a >5 Mbps internet connection.
General args:
- species Choice of 'homo_sapiens' or 'mus_musculus'. Default: 'homo_sapiens'.
- gene Str or list of gene name(s) or Ensembl ID(s), e.g. ['ACE2', 'SLC5A1'] or ['ENSG00000130234', 'ENSG00000100170']. Default: None.
NOTE: Set ensembl=True when providing Ensembl ID(s) instead of gene name(s).
See https://cellxgene.cziscience.com/gene-expression for examples of available genes.
- ensembl True/False (default: False). Set to True when genes are provided as Ensembl IDs.
- column_names List of metadata columns to return (stored in AnnData.obs when meta_only=False).
Default: ["dataset_id", "assay", "suspension_type", "sex", "tissue_general", "tissue", "cell_type"]
For more options see: https://api.cellxgene.cziscience.com/curation/ui/#/ -> Schemas -> dataset
- meta_only True/False (default: False). If True, returns only metadata dataframe (corresponds to AnnData.obs).
- census_version Str defining version of Census, e.g. "2023-05-15" or "latest" or "stable". Default: "stable".
- verbose True/False whether to print progress information. Default True.
- out If provided, saves the generated AnnData h5ad (or csv when meta_only=True) file with the specified path. Default: None.
Cell metadata attributes:
- tissue Str or list of tissue(s), e.g. ['lung', 'blood']. Default: None.
See https://cellxgene.cziscience.com/gene-expression for examples of available tissues.
- cell_type Str or list of celltype(s), e.g. ['mucus secreting cell', 'neuroendocrine cell']. Default: None.
See https://cellxgene.cziscience.com/gene-expression and select a tissue to see examples of available celltypes.
- development_stage Str or list of development stage(s). Default: None.
- disease Str or list of disease(s). Default: None.
- sex Str or list of sex(es), e.g. 'female'. Default: None.
- is_primary_data True/False (default: True). If True, returns only the canonical instance of the cellular observation.
This is commonly set to False for meta-analyses reusing data or for secondary views of data.
- dataset_id Str or list of CELLxGENE dataset ID(s). Default: None.
- tissue_general_ontology_term_id Str or list of high-level tissue UBERON ID(s). Default: None.
Also see: https://github.com/chanzuckerberg/single-cell-data-portal/blob/9b94ccb0a2e0a8f6182b213aa4852c491f6f6aff/backend/wmg/data/tissue_mapper.py
- tissue_general Str or list of high-level tissue label(s). Default: None.
Also see: https://github.com/chanzuckerberg/single-cell-data-portal/blob/9b94ccb0a2e0a8f6182b213aa4852c491f6f6aff/backend/wmg/data/tissue_mapper.py
- tissue_ontology_term_id Str or list of tissue ontology term ID(s) as defined in the CELLxGENE dataset schema. Default: None.
- assay_ontology_term_id Str or list of assay ontology term ID(s) as defined in the CELLxGENE dataset schema. Default: None.
- assay Str or list of assay(s) as defined in the CELLxGENE dataset schema. Default: None.
- cell_type_ontology_term_id Str or list of celltype ontology term ID(s) as defined in the CELLxGENE dataset schema. Default: None.
- development_stage_ontology_term_id Str or list of development stage ontology term ID(s) as defined in the CELLxGENE dataset schema. Default: None.
- disease_ontology_term_id Str or list of disease ontology term ID(s) as defined in the CELLxGENE dataset schema. Default: None.
- donor_id Str or list of donor ID(s) as defined in the CELLxGENE dataset schema. Default: None.
- self_reported_ethnicity_ontology_term_id Str or list of self reported ethnicity ontology ID(s) as defined in the CELLxGENE dataset schema. Default: None.
- self_reported_ethnicity Str or list of self reported ethnicity as defined in the CELLxGENE dataset schema. Default: None.
- sex_ontology_term_id Str or list of sex ontology ID(s) as defined in the CELLxGENE dataset schema. Default: None.
- suspension_type Str or list of suspension type(s) as defined in the CELLxGENE dataset schema. Default: None.
Returns AnnData object (when meta_only=False) or dataframe (when meta_only=True).
Fetch an AnnData object by selecting gene(s), tissue(s) and cell type(s)
You can use all of the options listed above to filter for data of interest. Here, we will demonstrate the module by fetching a small dataset containing only three genes and two lung cell types:
[3]:
# Fetch AnnData object based on specified genes, tissue and cell types
adata = gget.cellxgene(
gene=["ACE2", "ABCA1", "SLC5A1"], tissue="lung", cell_type=["mucus secreting cell", "neuroendocrine cell"]
)
Fri Jul 28 16:16:22 2023 INFO Fetching AnnData object from CZ CELLxGENE Discover. This might take a few minutes...
The "stable" release is currently 2023-07-25. Specify 'census_version="2023-07-25"' in future calls to open_soma() to ensure data consistency.
Fri Jul 28 16:16:22 2023 INFO The "stable" release is currently 2023-07-25. Specify 'census_version="2023-07-25"' in future calls to open_soma() to ensure data consistency.
Let’s look at some of the features of the AnnData object we just fetched:
[4]:
adata
[4]:
AnnData object with n_obs × n_vars = 3679 × 3
obs: 'dataset_id', 'assay', 'suspension_type', 'sex', 'tissue_general', 'tissue', 'cell_type', 'is_primary_data'
var: 'soma_joinid', 'feature_id', 'feature_name', 'feature_length'
A few thousand cells from CZ CELLxGENE Discover matched the filters specified above and their ACE2, ABCA1, and SLC5A1 expression matrix in lung mucus secreting and neuroendocrine cells was fetched. The .var and .obs layers contain additional information about each gene and cell, respectively:
[5]:
adata.var
[5]:
| soma_joinid | feature_id | feature_name | feature_length | |
|---|---|---|---|---|
| 0 | 38 | ENSG00000165029 | ABCA1 | 11343 |
| 1 | 5332 | ENSG00000130234 | ACE2 | 9739 |
| 2 | 24539 | ENSG00000100170 | SLC5A1 | 5081 |
[6]:
adata.obs
[6]:
| dataset_id | assay | suspension_type | sex | tissue_general | tissue | cell_type | is_primary_data | |
|---|---|---|---|---|---|---|---|---|
| 0 | 9f222629-9e39-47d0-b83f-e08d610c7479 | 10x 3' v2 | cell | unknown | lung | lung | mucus secreting cell | True |
| 1 | 9f222629-9e39-47d0-b83f-e08d610c7479 | 10x 3' v2 | cell | unknown | lung | lung | mucus secreting cell | True |
| 2 | 9f222629-9e39-47d0-b83f-e08d610c7479 | 10x 3' v2 | cell | unknown | lung | lung | mucus secreting cell | True |
| 3 | 9f222629-9e39-47d0-b83f-e08d610c7479 | 10x 3' v2 | cell | unknown | lung | lung | mucus secreting cell | True |
| 4 | 9f222629-9e39-47d0-b83f-e08d610c7479 | 10x 3' v2 | cell | unknown | lung | lung | mucus secreting cell | True |
| ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 3674 | 8c42cfd0-0b0a-46d5-910c-fc833d83c45e | 10x 3' v2 | cell | female | lung | lung | mucus secreting cell | True |
| 3675 | 8c42cfd0-0b0a-46d5-910c-fc833d83c45e | 10x 3' v2 | cell | female | lung | lung | mucus secreting cell | True |
| 3676 | 8c42cfd0-0b0a-46d5-910c-fc833d83c45e | 10x 3' v2 | cell | female | lung | lung | mucus secreting cell | True |
| 3677 | 8c42cfd0-0b0a-46d5-910c-fc833d83c45e | 10x 3' v2 | cell | female | lung | lung | mucus secreting cell | True |
| 3678 | 8c42cfd0-0b0a-46d5-910c-fc833d83c45e | 10x 3' v2 | cell | female | lung | lung | mucus secreting cell | True |
3679 rows × 8 columns
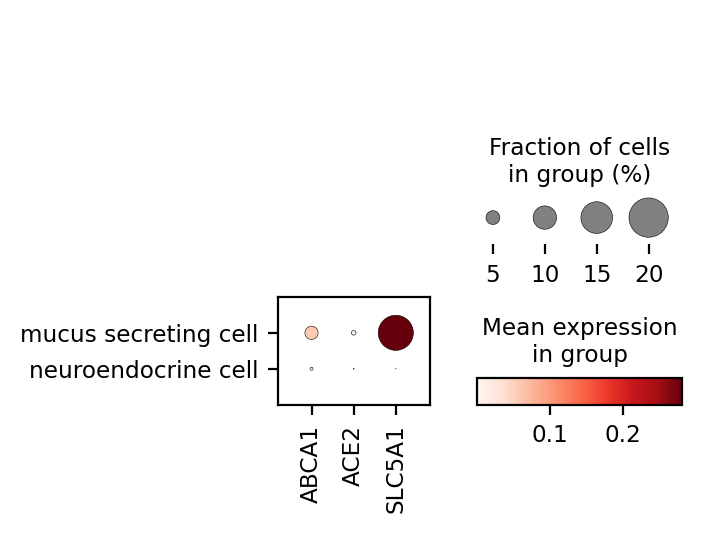
Plot a dot plot similar to those shown on the CZ CELLxGENE Discover Gene Expression
Using the data we just fetched, we can plot a dot plot using scanpy:
[7]:
import scanpy as sc
# retina increases the resolution of plots displayed in notebooks
%config InlineBackend.figure_format="retina"
[8]:
sc.pl.dotplot(adata, adata.var["feature_name"].values, groupby="cell_type", gene_symbols="feature_name")
... storing 'dataset_id' as categorical
... storing 'assay' as categorical
... storing 'suspension_type' as categorical
... storing 'sex' as categorical
... storing 'tissue_general' as categorical
... storing 'tissue' as categorical
... storing 'cell_type' as categorical
Fetch only cell metadata (corresponds to AnnData.obs)
By setting meta_only=True and again filtering by the cell metadata attributes listed above, you can also fetch only the cell metadata:
[9]:
df = gget.cellxgene(
meta_only=True,
census_version="2023-05-15", # Specify Census version for reproducibility over time
gene="ENSMUSG00000015405",
ensembl=True, # Setting 'ensembl=True' here since the gene is passed as an Ensembl ID
tissue="lung",
species="mus_musculus", # Let's switch up the species
)
df
Fri Jul 28 16:16:44 2023 INFO Fetching metadata from CZ CELLxGENE Discover...
[9]:
| dataset_id | assay | suspension_type | sex | tissue_general | tissue | cell_type | is_primary_data | |
|---|---|---|---|---|---|---|---|---|
| 0 | 047d57f2-4d14-45de-aa98-336c6f583750 | 10x 3' v2 | cell | unknown | lung | lung | mesenchymal stem cell | True |
| 1 | 047d57f2-4d14-45de-aa98-336c6f583750 | 10x 3' v2 | cell | unknown | lung | lung | progenitor cell | True |
| 2 | 047d57f2-4d14-45de-aa98-336c6f583750 | 10x 3' v2 | cell | unknown | lung | lung | mesenchymal cell | True |
| 3 | 047d57f2-4d14-45de-aa98-336c6f583750 | 10x 3' v2 | cell | unknown | lung | lung | mesenchymal stem cell | True |
| 4 | 047d57f2-4d14-45de-aa98-336c6f583750 | 10x 3' v2 | cell | unknown | lung | lung | mesenchymal cell | True |
| ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 97547 | 48b37086-25f7-4ecd-be66-f5bb378e3aea | 10x 3' v2 | cell | male | lung | lung | fibroblast of lung | True |
| 97548 | 48b37086-25f7-4ecd-be66-f5bb378e3aea | 10x 3' v2 | cell | male | lung | lung | natural killer cell | True |
| 97549 | 48b37086-25f7-4ecd-be66-f5bb378e3aea | 10x 3' v2 | cell | male | lung | lung | pulmonary interstitial fibroblast | True |
| 97550 | 48b37086-25f7-4ecd-be66-f5bb378e3aea | 10x 3' v2 | cell | male | lung | lung | adventitial cell | True |
| 97551 | 48b37086-25f7-4ecd-be66-f5bb378e3aea | 10x 3' v2 | cell | male | lung | lung | fibroblast of lung | True |
97552 rows × 8 columns
Use gget cellxgene from the command line
All gget modules support use from the command line. Note that the command line interface requires the -o/--out argument to specify a path to save the fetched data. Here are the command line versions of the queries demonstrated above:
[10]:
# # Fetch AnnData object based on specified genes, tissue and cell types
# !gget cellxgene --gene ACE2 ABCA1 SLC5A1 --tissue lung --cell_type 'mucus secreting cell' 'neuroendocrine cell' -o example_adata.h5ad
[11]:
# # Fetch only metadata
# !gget cellxgene --meta_only --gene ENSMUSG00000015405 --ensembl --tissue lung --species mus_musculus -o example_meta.csv