Train an scVI model using Census data
Authors: Emanuele Bezzi, Martin Kim, Mike Lin
This notebook demonstrates a scalable approach to training an scVI model on Census data. The scvi-tools library is built around PyTorch Lightning. TileDB-SOMA-ML assists with streaming Census query results to PyTorch in batches, allowing for training datasets larger than available RAM.
Contents
Training the model
Generate cell embeddings
Analyzing the results
Training the model
Let’s start by importing the necessary dependencies.
[1]:
import warnings
from typing import Any, Dict, List
import cellxgene_census
import numpy as np
import pandas as pd
import scanpy as sc
import scvi
import tiledbsoma as soma
import tiledbsoma_ml
import torch
from cellxgene_census.experimental.pp import highly_variable_genes
from lightning import LightningDataModule
from sklearn.preprocessing import LabelEncoder
from torch.utils.data import DataLoader
warnings.filterwarnings("ignore")
/opt/conda/lib/python3.11/site-packages/docrep/decorators.py:43: SyntaxWarning: 'param_categorical_covariate_keys' is not a valid key!
doc = func(self, args[0].__doc__, *args[1:], **kwargs)
/opt/conda/lib/python3.11/site-packages/docrep/decorators.py:43: SyntaxWarning: 'param_continuous_covariate_keys' is not a valid key!
doc = func(self, args[0].__doc__, *args[1:], **kwargs)
We’ll now prepare the necessary parameters for running a training pass of the model.
For this notebook, we’ll use a stable version of the Census:
[2]:
census = cellxgene_census.open_soma(census_version="2023-12-15")
We’ll also do two types of filtering.
For cells, we will apply a filter to only select primary cells, with at least 300 expressed genes (nnz >= 300). For notebook demonstration purposes, we will also apply a tissue filtering so that the training can happen on a laptop. The same approach can be used on datasets much larger than available RAM. (A GPU is recommended, though.)
For genes, we will apply a filter so that only the top 8000 highly variable genes (HVG) are included in the training. This is a commonly used dimensionality reduction approach and is recommended on production models as well.
Let’s define a few parameters:
[3]:
experiment_name = "mus_musculus"
obs_value_filter = 'is_primary_data == True and tissue_general in ["spleen", "kidney"] and nnz >= 300'
top_n_hvg = 8000
hvg_batch = ["assay", "suspension_type"]
For HVG, we can use the highly_variable_genes function provided in cellxgene_census, which can compute HVGs in constant memory:
[4]:
hvgs_df = highly_variable_genes(
census["census_data"][experiment_name].axis_query(
measurement_name="RNA", obs_query=soma.AxisQuery(value_filter=obs_value_filter)
),
n_top_genes=top_n_hvg,
batch_key=hvg_batch,
)
hv = hvgs_df.highly_variable
hv_idx = hv[hv].index
We will now introduce a helper class SCVIDataModule to connect TileDB-SOMA-ML with PyTorch Lightning. It subclasses LightningDataModule and:
Uses TileDB-SOMA-ML to prepare a DataLoader for the results of a SOMA ExperimentAxisQuery on the Census.
Derives each cell’s scVI batch label as a tuple of obs attributes:
dataset_id,assay,suspension_type,donor_id.Don’t confuse each cell’s label for scVI “batch” integration with a training data “batch” generated by the DataLoader.
Converts the RNA counts and batch labels to a dict of tensors for each training data batch, as scVI expects.
[5]:
class SCVIDataModule(LightningDataModule):
"""PyTorch Lightning DataModule for training scVI models from SOMA data
Wraps a `tiledbsoma_ml.ExperimentDataset` to stream the results of a SOMA `ExperimentAxisQuery`,
exposing a `DataLoader` to generate tensors ready for scVI model training. Also handles deriving
the scVI batch label as a tuple of obs columns.
"""
def __init__(
self,
query: soma.ExperimentAxisQuery,
*args,
batch_column_names: List[str] | None = None,
batch_labels: List[str] | None = None,
dataloader_kwargs: Dict[str, Any] | None = None,
**kwargs,
):
"""Args:
query: tiledbsoma.ExperimentAxisQuery
Defines the desired result set from a SOMA Expeirement.
*args, **kwargs:
Additional arguments passed through to `tiledbsoma_ml.ExperimentDataset`.
batch_column_names: List[str], optional
List of obs column names, the tuple of which defines the scVI batch label (not to to be confused with
a batch of training data). Defaults to
`["dataset_id", "assay", "suspension_type", "donor_id"]`.
batch_labels: List[str], optional
List of possible values of the batch label, for mapping to label tensors. By default,
this will be derived from the unique labels in the given query results (given
`batch_column_names`), making the label mapping depend on the query. The `batch_labels`
attribute in the `SCVIDataModule` used for training may be saved and here restored in
another instance for a different query. That ensures the label mapping will be correct
for the trained model, even if the second query doesn't return examples of every
training batch label.
dataloader_kwargs: dict, optional
Keyword arguments passed to `tiledbsoma_ml.experiment_dataloader()`, e.g. `num_workers`.
"""
super().__init__()
self.query = query
self.dataset_args = args
self.dataset_kwargs = kwargs
self.dataloader_kwargs = dataloader_kwargs if dataloader_kwargs is not None else {}
self.batch_column_names = (
batch_column_names
if batch_column_names is not None
else ["dataset_id", "assay", "suspension_type", "donor_id"]
)
self.batch_colsep = "//"
self.batch_colname = "scvi_batch"
# prepare LabelEncoder for the scVI batch label:
# 1. read obs DataFrame for the whole query result set
# 2. add scvi_batch column
# 3. fit LabelEncoder to the scvi_batch column's unique values
if batch_labels is None:
obs_df = self.query.obs(column_names=self.batch_column_names).concat().to_pandas()
self._add_batch_col(obs_df, inplace=True)
batch_labels = obs_df[self.batch_colname].unique()
self.batch_labels = batch_labels
self.batch_encoder = LabelEncoder().fit(self.batch_labels)
def setup(self, stage: str | None = None) -> None:
# Instantiate the ExperimentDataset with the provided args and kwargs.
self.train_dataset = tiledbsoma_ml.ExperimentDataset(
self.query, *self.dataset_args, obs_column_names=self.batch_column_names, **self.dataset_kwargs
)
def train_dataloader(self) -> DataLoader:
return tiledbsoma_ml.experiment_dataloader(
self.train_dataset,
**self.dataloader_kwargs,
)
def _add_batch_col(self, obs_df: pd.DataFrame, inplace: bool = False):
# synthesize a new column for obs_df by concatenating the self.batch_column_names columns
if not inplace:
obs_df = obs_df.copy()
obs_df[self.batch_colname] = obs_df[self.batch_column_names].astype(str).agg(self.batch_colsep.join, axis=1)
return obs_df
def on_before_batch_transfer(
self,
batch,
dataloader_idx: int,
) -> dict[str, torch.Tensor | None]:
# DataModule hook: transform the ExperimentDataset data batch (X: ndarray, obs_df: DataFrame)
# into X & batch variable tensors for scVI (using batch_encoder on scvi_batch)
batch_X, batch_obs = batch
self._add_batch_col(batch_obs, inplace=True)
return {
"X": torch.from_numpy(batch_X).float(),
"batch": torch.from_numpy(self.batch_encoder.transform(batch_obs[self.batch_colname])).unsqueeze(1),
"labels": torch.empty(0),
}
# scVI code expects these properties on the DataModule:
@property
def n_obs(self) -> int:
return len(self.query.obs_joinids())
@property
def n_vars(self) -> int:
return len(self.query.var_joinids())
@property
def n_batch(self) -> int:
return len(self.batch_encoder.classes_)
[6]:
hvg_query = census["census_data"][experiment_name].axis_query(
measurement_name="RNA",
obs_query=soma.AxisQuery(value_filter=obs_value_filter),
var_query=soma.AxisQuery(coords=(list(hv_idx),)),
)
datamodule = SCVIDataModule(
hvg_query,
layer_name="raw",
batch_size=1024,
shuffle=True,
seed=42,
dataloader_kwargs={"num_workers": 0, "persistent_workers": False},
)
(datamodule.n_obs, datamodule.n_vars, datamodule.n_batch)
[6]:
(203655, 8000, 43)
Most parameters to SCVIDataModule are passed through to the tiledbsoma_ml.ExperimentDataset initializer; see that documentation to understand how it can be tuned.
In particular, here are some parameters of interest:
shuffle: shuffles the result cell order, which is often advisable for model training.batch_size: controls the size (number of cells) in each training data batch, in turn controlling memory usage.dataloader_kwargs: DataLoader tuning, for example controlling parallelization.
We can now create the scVI model object:
[7]:
n_layers = 1
n_latent = 50
model = scvi.model.SCVI(n_layers=n_layers, n_latent=n_latent, gene_likelihood="nb", encode_covariates=False)
Then, we can invoke the .train method which will start the training loop. For this demonstration, we’ll only do a single epoch, but this should likely be increased for a production model. The scVI models hosted in CELLxGENE have been trained for 100 epochs.
[8]:
model.train(
datamodule=datamodule,
max_epochs=1,
batch_size=1024,
train_size=0.9,
early_stopping=False,
)
GPU available: True (cuda), used: True
TPU available: False, using: 0 TPU cores
HPU available: False, using: 0 HPUs
You are using a CUDA device ('NVIDIA A10G') that has Tensor Cores. To properly utilize them, you should set `torch.set_float32_matmul_precision('medium' | 'high')` which will trade-off precision for performance. For more details, read https://pytorch.org/docs/stable/generated/torch.set_float32_matmul_precision.html#torch.set_float32_matmul_precision
LOCAL_RANK: 0 - CUDA_VISIBLE_DEVICES: [0]
`Trainer.fit` stopped: `max_epochs=1` reached.
We can now save the trained model. As of the current writing, scvi-tools doesn’t support saving a model that wasn’t generated through an AnnData loader, so we’ll use some custom code:
[9]:
model_state_dict = model.module.state_dict()
var_names = hv_idx.to_numpy()
user_attributes = model._get_user_attributes()
user_attributes = {a[0]: a[1] for a in user_attributes if a[0][-1] == "_"}
user_attributes.update(
{
"n_batch": datamodule.n_batch,
"n_extra_categorical_covs": 0,
"n_extra_continuous_covs": 0,
"n_labels": 1,
"n_vars": datamodule.n_vars,
"batch_labels": datamodule.batch_labels,
}
)
with open("model.pt", "wb") as f:
torch.save(
{
"model_state_dict": model_state_dict,
"var_names": var_names,
"attr_dict": user_attributes,
},
f,
)
We will now load the model back and use it to generate cell embeddings (the latent space), which can then be used for further analysis. Loading the model similarly involves some custom code.
[10]:
with open("model.pt", "rb") as f:
torch_model = torch.load(f)
adict = torch_model["attr_dict"]
params = adict["init_params_"]["non_kwargs"]
n_batch = adict["n_batch"]
n_extra_categorical_covs = adict["n_extra_categorical_covs"]
n_extra_continuous_covs = adict["n_extra_continuous_covs"]
n_labels = adict["n_labels"]
n_vars = adict["n_vars"]
latent_distribution = params["latent_distribution"]
dispersion = params["dispersion"]
n_hidden = params["n_hidden"]
dropout_rate = params["dropout_rate"]
gene_likelihood = params["gene_likelihood"]
model = scvi.model.SCVI(
n_layers=params["n_layers"],
n_latent=params["n_latent"],
gene_likelihood=params["gene_likelihood"],
encode_covariates=False,
)
module = model._module_cls(
n_input=n_vars,
n_batch=n_batch,
n_labels=n_labels,
n_continuous_cov=n_extra_continuous_covs,
n_cats_per_cov=None,
n_hidden=n_hidden,
n_latent=n_latent,
n_layers=n_layers,
dropout_rate=dropout_rate,
dispersion=dispersion,
gene_likelihood=gene_likelihood,
latent_distribution=latent_distribution,
)
model.module = module
model.module.load_state_dict(torch_model["model_state_dict"])
device = torch.device("cuda:0" if torch.cuda.is_available() else "cpu")
model.to_device(device)
model.module.eval()
model.is_trained = True
Generate cell embeddings
We will now generate the cell embeddings for this model, using the get_latent_representation function available in scvi-tools.
We can use another instance of the SCVIDataModule for the forward pass, so we don’t need to load the whole dataset in memory. This will have shuffling disabled to make it easier to join the embeddings later. We also want to restore the list of scVI batch labels from the training data, ensuring our forward pass will map batch labels to tensors in the expected way (although this specific example would work regardless, since it reuses the same query).
[11]:
inference_datamodule = SCVIDataModule(
hvg_query,
layer_name="raw",
batch_labels=adict["batch_labels"],
batch_size=1024,
shuffle=False,
dataloader_kwargs={"num_workers": 0, "persistent_workers": False},
)
To feed the data to get_latent_representation, we operate inference_datamodule as PyTorch Lightning would during training:
[12]:
inference_datamodule.setup()
inference_dataloader = (
inference_datamodule.on_before_batch_transfer(batch, None) for batch in inference_datamodule.train_dataloader()
)
latent = model.get_latent_representation(dataloader=inference_dataloader)
latent.shape
[12]:
(203655, 50)
We successfully trained the model and generated embeddings using limited memory. Even on the full Census, this has been tested to run with less than 30G of memory.
Analyzing the results
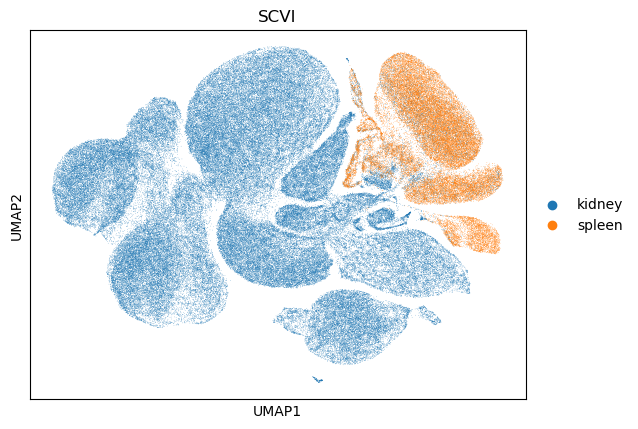
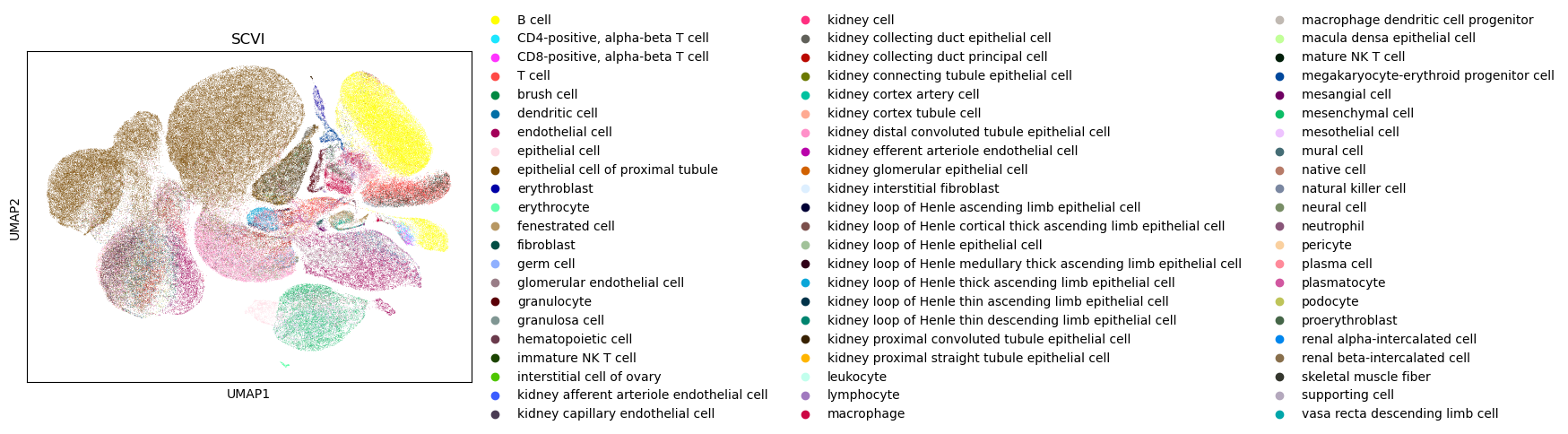
We will now take a look at the UMAP for the generated embedding. Note that this model was only trained for one epoch (for demo purposes), so we don’t expect the UMAP to show significant integration patterns, but it is nonetheless a good way to check the overall health of the generated embedding.
In order to do this, we’ll use scanpy which accepts an AnnData object, so we’ll generate one using the get_anndata utility function:
[13]:
adata = cellxgene_census.get_anndata(
census,
organism=experiment_name,
obs_value_filter=obs_value_filter,
)
Add the generated embedding (stored in latent) in the obsm slot of the AnnData object:
[14]:
# verify cell order:
assert np.array_equal(np.array(adata.obs["soma_joinid"]), inference_datamodule.train_dataset.query_ids.obs_joinids)
adata.obsm["scvi"] = latent
We can now generate the neighbors and the UMAP.
[ ]:
sc.pp.neighbors(adata, use_rep="scvi", key_added="scvi")
sc.tl.umap(adata, neighbors_key="scvi")
sc.pl.umap(adata, color="dataset_id", title="SCVI")
2025-03-09 23:51:44.271925: E external/local_xla/xla/stream_executor/cuda/cuda_fft.cc:485] Unable to register cuFFT factory: Attempting to register factory for plugin cuFFT when one has already been registered
2025-03-09 23:51:44.285009: E external/local_xla/xla/stream_executor/cuda/cuda_dnn.cc:8454] Unable to register cuDNN factory: Attempting to register factory for plugin cuDNN when one has already been registered
2025-03-09 23:51:44.288982: E external/local_xla/xla/stream_executor/cuda/cuda_blas.cc:1452] Unable to register cuBLAS factory: Attempting to register factory for plugin cuBLAS when one has already been registered

[ ]:
sc.pl.umap(adata, color="tissue_general", title="SCVI")
[ ]:
sc.pl.umap(adata, color="cell_type", title="SCVI")
[ ]: