Normalizing full-length gene sequencing data
This tutorial shows you how to fetch full-length gene sequencing data from the Census and normalize it to account for gene length.
Contents
Opening the census
Fetching example full-length sequencing data (Smart-Seq2)
Normalizing expression to account for gene length
Validation through clustering exploration
⚠️ Note that the Census RNA data includes duplicate cells present across multiple datasets. Duplicate cells can be filtered in or out using the cell metadata variable is_primary_data which is described in the Census schema. For this notebook we will focus on individual datasets, therefore we can ignore this variable.
Opening the census
First we open the Census, if you are not familiar with the basics of the Census API you should take a look at notebook Learning about the CZ CELLxGENE Census
[1]:
import cellxgene_census
import scanpy as sc
from scipy.sparse import csr_matrix
census = cellxgene_census.open_soma()
The "stable" release is currently 2023-07-25. Specify 'census_version="2023-07-25"' in future calls to open_soma() to ensure data consistency.
You can learn more about the all of the cellxgene_census methods by accessing their corresponding documention via help(). For example help(cellxgene_census.open_soma).
Fetching full-length example sequencing data (Smart-Seq)
Let’s get some example data, in this case we’ll fetch all cells from a relatively small dataset derived from the Smart-Seq2 technology which performs full-length gene sequencing:
Collection: Tabula Muris Senis
Dataset: Liver - A single-cell transcriptomic atlas characterizes ageing tissues in the mouse - Smart-seq2
Let’s first find this dataset’s id by using the dataset table of the Census
[2]:
liver_dataset = (
census["census_info"]["datasets"]
.read(
value_filter="dataset_title == 'Liver - A single-cell transcriptomic atlas characterizes ageing tissues in the mouse - Smart-seq2'"
)
.concat()
.to_pandas()
)
liver_dataset
[2]:
| soma_joinid | collection_id | collection_name | collection_doi | dataset_id | dataset_title | dataset_h5ad_path | dataset_total_cell_count | |
|---|---|---|---|---|---|---|---|---|
| 0 | 525 | 0b9d8a04-bb9d-44da-aa27-705bb65b54eb | Tabula Muris Senis | 10.1038/s41586-020-2496-1 | 4546e757-34d0-4d17-be06-538318925fcd | Liver - A single-cell transcriptomic atlas cha... | 4546e757-34d0-4d17-be06-538318925fcd.h5ad | 2859 |
Now we can use this id to fetch the data.
[3]:
liver_dataset_id = "4546e757-34d0-4d17-be06-538318925fcd"
liver_adata = cellxgene_census.get_anndata(
census,
organism="Mus musculus",
obs_value_filter=f"dataset_id=='{liver_dataset_id}'",
)
Let’s make sure this data only contains Smart-Seq2 cells
[4]:
liver_adata.obs["assay"].value_counts()
[4]:
assay
Smart-seq2 2859
Name: count, dtype: int64
Great! As you can see this a small dataset only containing 2,859 cells. Now let’s proceed to normalize by gene lengths.
For good practice let’s add the number of genes expressed by cell, and the total counts per cell.
Don’t forget to close the census
[5]:
census.close()
Normalizing expression to account for gene length
By default cellxgene_census.get_anndata() fetches all genes in the Census. So let’s first identify the genes that were measured in this dataset and subset the AnnData to only include those.
To this goal we can use the “Dataset Presence Matrix” in census["census_data"]["mus_musculus"].ms["RNA"]["feature_dataset_presence_matrix"]. This is a boolean matrix N x M where N is the number of datasets and M is the number of genes in the Census, True indicates that a gene was measured in a dataset.
[6]:
liver_adata.n_vars
[6]:
52392
Let’s get the genes measured in this dataset.
[7]:
liver_dataset_id = liver_dataset["soma_joinid"][0]
presence_matrix = cellxgene_census.get_presence_matrix(census, "Mus musculus", "RNA")
presence_matrix = presence_matrix[liver_dataset_id, :]
gene_presence = presence_matrix.nonzero()[1]
liver_adata = liver_adata[:, gene_presence].copy()
[8]:
liver_adata.n_vars
[8]:
17992
We can see that out of the all genes in the Census 17,992 were measured in this dataset.
Now let’s normalize these genes by gene length. We can easily do this because the Census has gene lengths included in the gene metadata under feature_length.
[9]:
liver_adata.X[:5, :5].toarray()
[9]:
array([[0.000e+00, 0.000e+00, 0.000e+00, 0.000e+00, 0.000e+00],
[0.000e+00, 0.000e+00, 5.590e+02, 0.000e+00, 0.000e+00],
[0.000e+00, 0.000e+00, 1.969e+03, 0.000e+00, 8.280e+02],
[0.000e+00, 0.000e+00, 0.000e+00, 0.000e+00, 1.000e+00],
[0.000e+00, 2.250e+03, 0.000e+00, 0.000e+00, 5.400e+01]],
dtype=float32)
[10]:
gene_lengths = liver_adata.var[["feature_length"]].to_numpy()
liver_adata.X = csr_matrix((liver_adata.X.T / gene_lengths).T)
[11]:
liver_adata.X[:5, :5].toarray()
[11]:
array([[0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
0.00000000e+00],
[0.00000000e+00, 0.00000000e+00, 6.58654413e-02, 0.00000000e+00,
0.00000000e+00],
[0.00000000e+00, 0.00000000e+00, 2.32001885e-01, 0.00000000e+00,
2.74444813e-01],
[0.00000000e+00, 0.00000000e+00, 0.00000000e+00, 0.00000000e+00,
3.31455088e-04],
[0.00000000e+00, 4.71500419e-01, 0.00000000e+00, 0.00000000e+00,
1.78985747e-02]])
All done! You can see that we now have real numbers instead of integers.
Validation through clustering exploration
Let’s perform some basic clustering analysis to see if cell types cluster as expected using the normalized counts.
First we do some basic filtering of cells and genes.
[12]:
sc.pp.filter_cells(liver_adata, min_genes=500)
sc.pp.filter_genes(liver_adata, min_cells=5)
Then we normalize to account for sequencing depth and transform data to log scale.
[13]:
sc.pp.normalize_total(liver_adata, target_sum=1e4)
sc.pp.log1p(liver_adata)
Then we subset to highly variable genes.
[14]:
sc.pp.highly_variable_genes(liver_adata, n_top_genes=1000)
liver_adata = liver_adata[:, liver_adata.var.highly_variable].copy()
And finally we scale values across the gene axis.
[15]:
sc.pp.scale(liver_adata, max_value=10)
Now we can proceed to do clustering analysis
[16]:
sc.tl.pca(liver_adata)
sc.pp.neighbors(liver_adata)
sc.tl.umap(liver_adata)
sc.pl.umap(liver_adata, color="cell_type")
/home/ssm-user/cellxgene-census/venv/lib/python3.10/site-packages/tqdm/auto.py:21: TqdmWarning: IProgress not found. Please update jupyter and ipywidgets. See https://ipywidgets.readthedocs.io/en/stable/user_install.html
from .autonotebook import tqdm as notebook_tqdm
/home/ssm-user/cellxgene-census/venv/lib/python3.10/site-packages/scanpy/plotting/_tools/scatterplots.py:392: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored
cax = scatter(
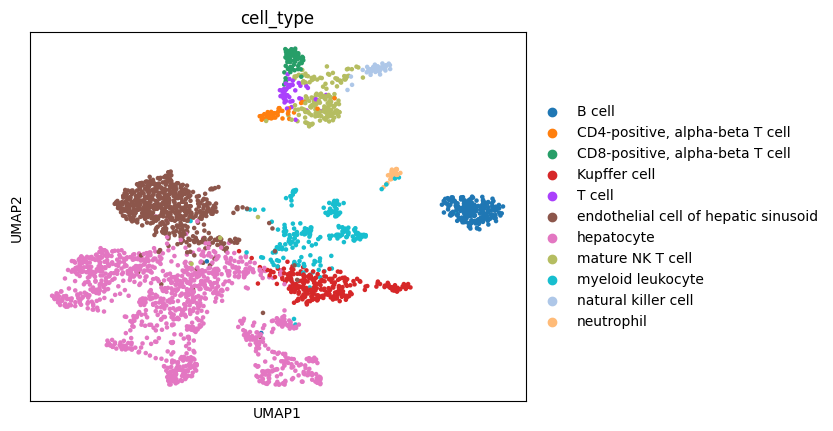
With a few exceptions we can see that all cells from the same cell type cluster near each other which serves as a sanity check for the gene-length normalization that we applied.